Our previous blog posts covered the history of the laws in the United States that govern the approval of therapeutics: the Federal Food, Drug, and Cosmetic Act (FDCA) of 1938 and the Public Health Service Act (PHSA) of 1944. We also previously covered how both laws were later amended to create, among other things, abbreviated pathways to approval. In the U.S., there are five different approval pathways for therapeutics.
FDCA or PHSA
Certain therapeutics are approved under the FDCA by the submission of a New Drug Application (NDA). Other therapeutics are approved under the PHSA by the submission of a Biological License Application (BLA). Whether a therapeutic is approved under an NDA or a BLA depends on the composition of the therapeutic and how it is manufactured. Our previous blog post “2023 Year in Review: approved NDAs” covered the therapeutics approved in 2023 under an NDA, which included small molecules, peptides, and oligonucleotides. Our blog post “2023 Year in Review: approved BLAs” covered the therapeutics approved in 2023 under a BLA which included antibodies, BiTEs, fusion proteins, small proteins, source plasma, cell therapy, vaccines, gene therapy and microbiome.
Thus, FDA approval for therapeutics such as small molecules, peptides and oligonucleotides fall under the FDCA and are approved under an NDA. Other therapeutics such as biological therapeutics (antibodies, BiTEs, fusion proteins, small proteins, source plasma, cell therapy, vaccines, gene therapy and microbiome) are approved under a BLA under the PHSA. Below is a diagram summarizing this information:
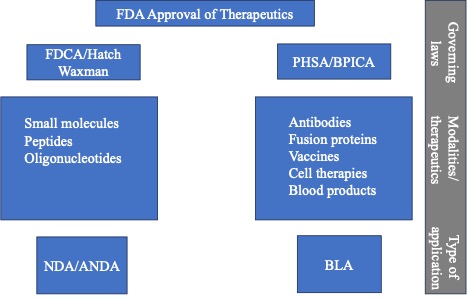
FDCA/Hatch-Waxman
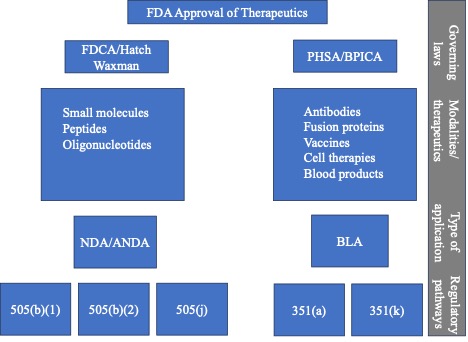
The Federal Food, Drug, and Cosmetic Act (FDCA) of 1938 was amended by the “Drug Price Competition and Patent Term Restoration Act of 1984,” also known as the Hatch-Waxman Amendments to increase generic drug entry. Under the FDCA, therapeutics are approved under section 505(b)(1) and applicants have to submit full NDAs. The Hatch-Waxman Amendments formalized two abbreviated pathways for therapeutic approvals by adding sections 505(b)(2) and 505(j). Thus, for therapeutics approved under the FDCA/Hatch-Waxman amendments, there are three approval pathways: 505(b)(1), 505(b)(2), and 505(j).
For any therapeutic to be approved under the FDCA via an NDA, the therapeutic must be:
- safe; and
- effective for the proposed use and the benefits outweigh the risks
Additionally, the manufacturing methods of the therapeutic must preserve the therapeutic’s identity, strength, quality, and purity.
To prove that the therapeutic is safe, effective, and that the benefits outweigh the risks, an applicant submits data to the FDA. This data package includes pre-clinical data, clinical data, and manufacturing data. The regulatory pathway for a therapeutic depends on the type of data available for that therapeutic.
505(b)(1)
Applications for therapeutics approved under 505(b)(1) are considered full-blown applications. Usually, the data submitted in an application under 505(b)(1) is obtained by the applicant by conducing pre-clinical and clinical studies. Meaning, the applicant designs, oversees and pays for the pre-clinical experiments and the clinical trials to get the data needed to prove to the FDA that the therapeutic is safe and effective. Sometimes, the applicant has not performed the experiments themselves, but has obtained a right of reference. With a right reference, the applicant has the right to use data that is owned by another entity. This may occur when an applicant has licensed a therapeutic from another company and as part of the license agreement has obtain the right to the data.
Thus, under 505(b)(1), the applicant obtains all the data, either by owning the data or by having a right of reference, that is needed to support the application to show that the therapeutic is safe and effective.
505(b)(2)
Applications for therapeutics approved under 505(b)(2) are not exactly full-blown applications, but are abbreviated applications. For example, in this situation there may exist data from previous studies where the applicant does not have a right of reference. Or there could be information in the scientific literature that the applicant relies upon to show to the FDA that the therapeutic is safe and effective. Instead of having to repeat these experiments, the applicant is able to rely upon previous work done by others to show to the FDA that the therapeutic is safe and effective.
Thus, under 505(b)(2), the applicant partially relies upon the data produced by others to prove to the FDA that the therapeutic is safe and effective.
505(j)
In certain situations, an applicant seeks approval from the FDA to market and commercialize a therapeutic that has already been approved by the FDA. Therapeutics that are copies of an already approved drug are considered a generic drug or generic therapeutic. The first or original approved therapeutic is often referred to as the innovator or brand therapeutic. It is also called the reference listed drug (RLD) by the FDA. To avoid an applicant having to repeat pre-clinical and clinical studies wth the RLD, the applicant can submit an application under 505(j). This is considered an abbreviated new drug application or ANDA. An “ANDA” or “505(j) application” copies the safety and efficacy data from an already approved therapeutic, called the reference drug, or RLD.
PHSA and BPCIA
Certain therapeutics that are considered biologicals are approved under a Biological License Application (BLA) under the PHSA. The PHSA was amended through the passage of the Biologics Price Competition and Innovation Act of 2009 (BPCIA). This legislation created the statutory authority for the FDA to approve generic versions of biological therapeutics, called biosimilars and interchangeables. Thus, with the PHSA and BPCIA, there are two pathways to approval for a biological therapeutic: 351(a) and 351(k).
For a biological therapeutic to be approved under a BLA, it must be shown to be:
- safe
- potent
- pure
As extensively covered in our Therapeutics and Modalities course, biological therapeutics are manufactured from cells. Because biological therapeutics are made from living systems there are tighter controls over their manufacturing processes to ensure that the product is pure, or does not contain impurities. To show that a biological therapeutic is safe, potent, and pure, an applicant submits pre-clinical and clinical data to the FDA. The pathway selected for a biological therapeutic is dependent on the type of data available.
351(a)
Applications for biological therapeutics submitted under 351(a) are full-blown applications. An application submitted under 351(a) contains all the information and data necessary to show that the biological therapeutic is safe, pure and potent. An applicant submits pre-clinical, clinical and manufacturing data to meet these requirements. A biological therapeutic approved under 351(a) is often referred to as an innovator drug, a brand drug, a reference drug or a reference product.
351(k)
In contrast, an application submitted under 351(k) needs to demonstrate that the biological therapeutic is a biosimilar to, or interchangeable with, a reference drug or a reference product (a biological therapeutic already approved under 351(a)). For a biological therapeutic to be a biosimilar to a reference product, it must be:
- highly similar to the reference product notwithstanding minor differences in clinically inactive components; and
- have no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency
For a biological therapeutic to be interchangeable with a reference product means that the biological therapeutic may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product. To be considered an interchangeable, it must be:
- a biosimilar to the reference product;
- expected to produce the same clinical result as the reference product in any given patient; and
- for a product that is administered more than once, the risk in terms of safety or diminished efficacy of alternating or switching between use of the product and the reference product is not greater than the risk of using the reference product
Only if the biological therapeutic meets these additional requirements can it be considered an interchangeable.
Thus, there are five approval pathways for therapeutics in the United States. The pathway selected depends on the type of therapeutic (small molecule, peptides, oligonucleotides, antibodies, BiTEs, fusion proteins, small proteins, source plasma, cell therapy, vaccines, gene therapy and microbiome) and the source of the data that will be used to support the NDA or BLA.
Additional resources
Understanding regulatory pathways is important for any professional working in the life science or biotech industry. Additionally, an understanding of the various modalities common in life sciences is also required no matter your particular role. Science for Bankers offers an Executive Course on Therapeutics and Modalities that covers each modality in depth. The course covers the five main classes of therapeutics, and much more. It is made up of about 45 videos; each under 30 minutes. Watch whenever and wherever. Essentially, a masterclass of therapeutics and modalities, or the science in life sciences.