April 9, 2025
Data exclusivity keeps drug competitors out of the market and preserves a company’s market share and revenue for a certain period of time. Data exclusivity only applies to certain therapeutics that have been approved by the Food and Drug Administration (FDA).
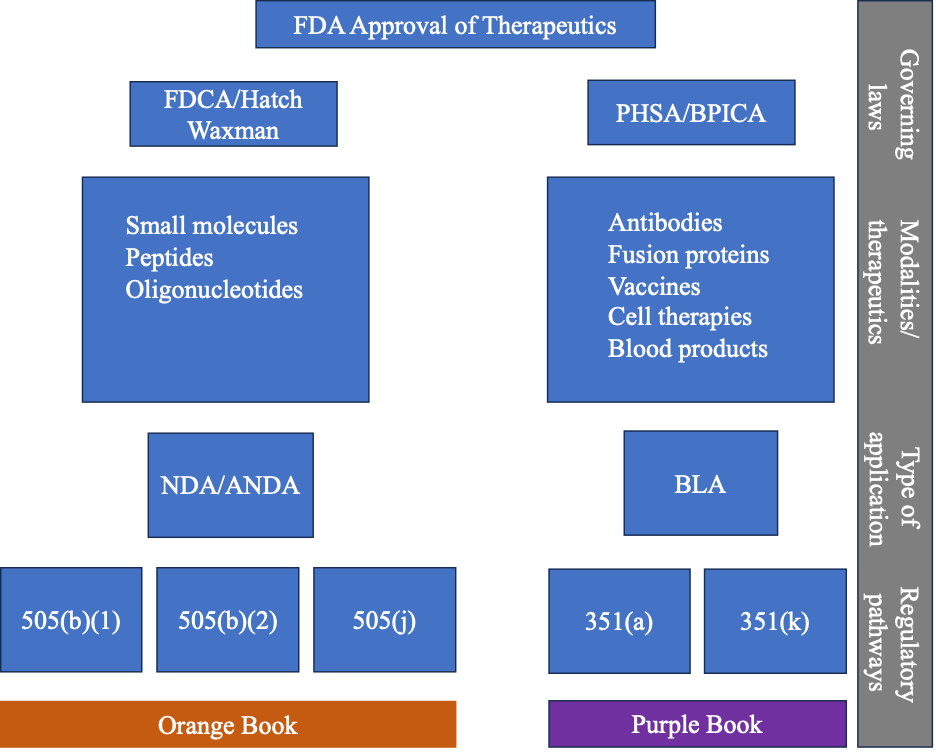
Before a drug or therapeutic can be commercialized and marketed in the United States, the therapeutic must be approved by the FDA. Whether the therapeutic is a small molecule, oligonucleotide, gene therapy, large molecule, ASO, siRNA, or aptamer, it must first be approved under either a new drug application (NDA) or biological license application (BLA).
Our previous blog posts covered the history of the laws that have led to the various approval pathways in the United States. We have covered the Federal Food, Drug, and Cosmetic Act (FDCA) of 1938, the Public Health Service Act (PHSA) in 1944, the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Amendments, and the Biologics Price Competition and Innovation Act of 2009 (BPCIA). For therapeutics approved under the FDCA/Hatch-Waxman via an NDA, there are three approval pathways: 505(b)(1), 505(b)(2), and 505(j). For therapeutics approved under the PHSA/BPCIA via a BLA, there are two pathways to approval for a biological therapeutic: 351(a) and 351(k).
Besides blog posts, Science for Bankers’ premium content includes videos on the regulatory pathways at the FDA in our “FDA School.” This content provides a series of videos about how the different therapeutics are reviewed and approved in the U.S., along with what regulatory exclusivities apply. The FDA School content can be accessed with a subscription to Science for Bankers’ premium content. Science for Bankers’ premium content also covers the major classes of modalities and therapeutics: large molecules, small molecules, gene therapy, cell therapy, and RNA based therapeutics.
The regulatory pathway is dictated by the type of therapeutic (whether it is approved under a BLA or NDA) and the availability of the data needed to get the therapeutic approved by the FDA. For a therapeutic to be approved under the FDCA via an NDA, the therapeutic must be:
- safe and
- effective for the proposed use
Additionally, the manufacturing methods of the therapeutic must preserve the therapeutic’s identity, strength, quality, and purity.
For a biological therapeutic to be approved under a BLA, it must be shown to be:
- safe;
- potent; and
- pure
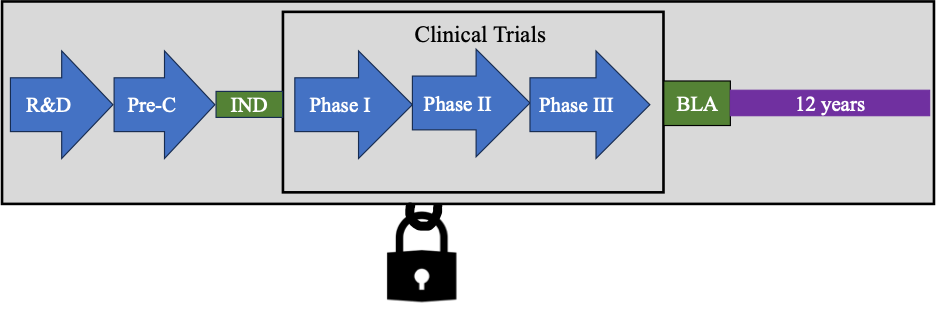
Once approved by the FDA under an NDA or BLA, information about the therapeutic is listed in either the Orange Book or the Purple Book. Both of these “books” are valuable online resources for approved therapeutics. The information covered in our previous blog posts can be summarized in the following diagram.
Getting a therapeutic approved by the FDA is an expensive, difficult, and time-consuming process. To demonstrate to the FDA that a therapeutic is safe and effective in an NDA or safe, potent, and pure under a BLA, data must be submitted to the FDA. Data is generated by conducting experiments and clinical trials, which require time, money, and resources to procure.
Research and Development
Let’s consider the example of a novel therapeutic; one that has never existed before. Usually through research and development (R&D) a therapeutic, such as a small molecule or an antibody, is discovered. R&D efforts can be based upon finding a particular therapeutic that binds or modulates a particular target in the body, with the theory that binding this target will treat a particular disease.
Pre-clinical Investigations
Therapeutics that are found to bind or modulate a target in in vitro experiments (for example, in cell-based assays) may be further investigated in in vivo studies (for example, in animal models). If the therapeutic is believed to be safe and effective in animal models, the company may decide to further the investigation in clinical trials. A data package comprising these pre-clinical experiments and investigations is submitted to the FDA in an investigational new drug (IND) application. The IND contains an extensive data package demonstrating the safety and efficacy of the therapeutic from pre-clinical studies. In the U.S., clinical trials can only begin once the IND becomes effective.
Clinical Investigations
Once the IND is effective, a company can begin clinical trials where the therapeutic is tested in humans for the first time. In a series of experiments, usually called phases (for example, phase I, phase II, and phase III), the therapeutic is tested in humans. If the therapeutic is successful in the clinic (shown to be safe and effective), an applicant may submit an extensive data package to the FDA in either an NDA or BLA. A diagram summarizing this process is shown below.
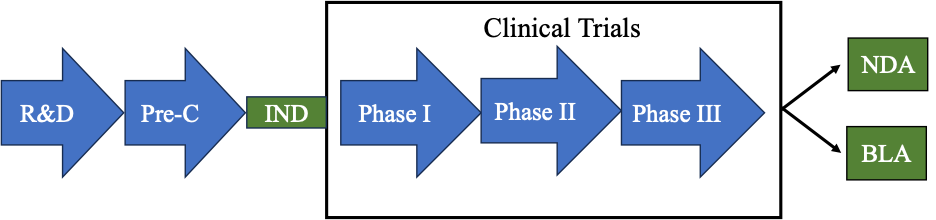
Obtaining the data to be submitted in a data package for an IND, NDA, or BLA submission is expensive and time consuming. There have been many studies that have investigated just how long and how expensive this process is for a company. On average it can take 10 – 15 years from R&D to NDA or BLA approval and the average cost has been estimated at $1.3 billion.
Needless to say, the data obtained during R&D, pre-clinical experiments and clinical trials is incredibly valuable to a company. Even though this data is submitted to the FDA in an IND, NDA or BLA, the FDA only discloses the data in rare circumstances. This data is highly confidential and can be considered a company’s trade secret. Most companies safeguard this information from public disclosure and may only disclose portions of the data in abstracts, posters, publications, and presentations.
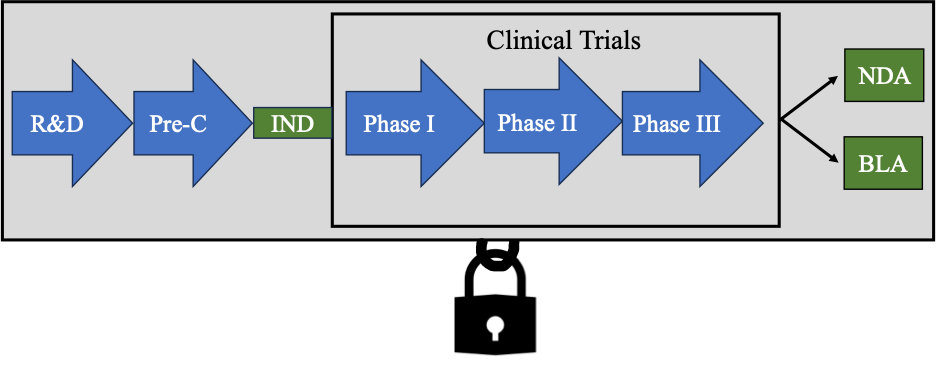
Data Exclusivity
Congress and the FDA recognized how valuable this data is to an applicant. To allow another company to copy this data and gain approval for a generic version of the therapeutic would allow the generic applicant an unfair advantage; to commercialize a drug without having to expend considerable resources such as time and money to get the therapeutic on the market. It would be unfair to let other companies rely on this data without cost.
However, generic versions of drugs can lower drug prices. To strike a balance between protecting the data obtained by innovators and yet encouraging generic drug entry, this data is “protected” for a certain period of time.
This is considered “data exclusivity.” Where an applicant’s data cannot be relied upon by other applicants for a certain period of time. It provides a form of market exclusivity. Data exclusivity usually means two things: (1) protecting preclinical and clinical data from public disclosure by the FDA, and (2) preventing other applicants from relying on the data for a period of time.
Data exclusivity time periods
Hatch-Waxman Amendments
Congress addressed data exclusivity in the Hatch-Waxman Amendments. Under the Hatch-Waxman Amendments, Congress provided new drug applicants with either three or five years of data exclusivity. The five year data exclusivity is referred to as NCE (new chemical entity exclusivity). The three year data exclusivity is referred to as NCI (new clinical investigation).
If an approved drug product contains no active moiety that has been approved by FDA in any other application submitted under section 505(b), then the drug product is granted five years of data exclusivity, or NCE.
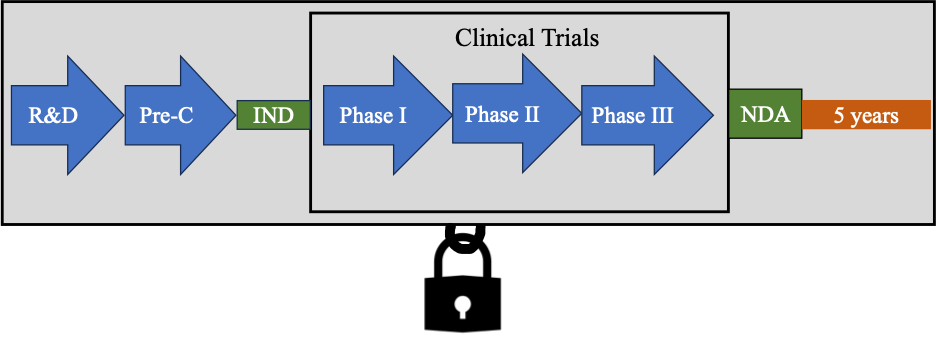
That means that an application submitted under 505(b)(2) or 505 (j) (also called an ANDA) that relies upon the applicant’s data may not be submitted during the 5-year exclusivity period. There is an exception that such applications may be submitted after 4 years if they contain a certification of patent invalidity or noninfringement. Because a 505(b)(2) or ANDA applicant cannot use the data for 5 years (data exclusivity), this creates a market exclusivity. During this time there is no competing product in the market.
Also, under the Hatch-Waxman Amendments, if an application for a proposed drug product requires a clinical investigation (other than bioavailability data), then the data is protected for three years. A competitor may not rely upon this data for three years, which provides a market exclusivity.
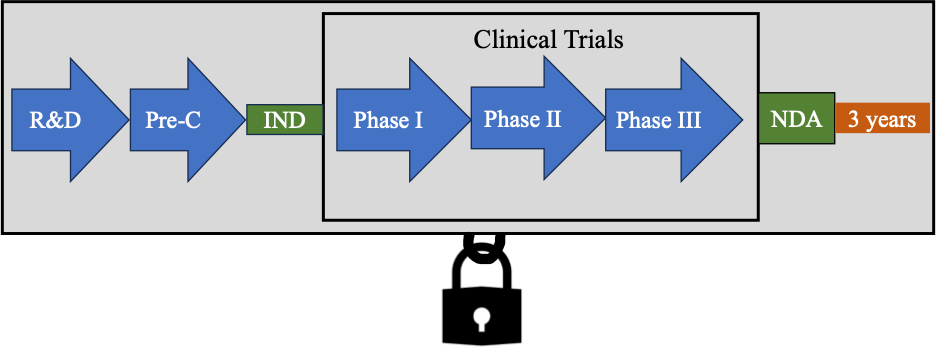
For example, if an applicant changed a drug product with regards to the active ingredient(s) strength, dosage form, route of administration or additional uses, and these changes necessitated clinical investigations to support the application, then the approved drug product would be granted three years of data exclusivity (NCI). In essence, if an applicant had to expend considerable time and money to gain the data needed for the approval, the drug product would be entitled to three years of data exclusivity.
Biologics Price Competition and Innovation Act of 2009 (BPCIA)
Under the BPCIA, a biological therapeutic approved under 351(a) is granted market exclusivity for 12 years. There are two key time points to keep in mind with regards to the BPCIA: 4 years and 12 years. For four years, a 351(k) application cannot be submitted for review that relies upon the applicant’s data. After 4 years, a 351(k) application can be submitted for review, but it cannot be approved until 12 years after approval. So, essentially, a biological therapeutic approved under 351(a) is granted 12 years of market exclusivity. Meaning that a biosimilar of the drug cannot be approved during this time.
Orphan Drug Exclusivity (ODE)
Under the 1983 Orphan Drug Act, therapeutics approved to treat rare diseases can get seven years of market exclusivity. Orphan drug exclusivity blocks the agency from approving generic versions of a drug for the same orphan indication during the exclusivity period.
Pediatric Exclusivity (PED)
Under the FDA Modernization Act of 1997, certain applicants are able to obtain six-month extensions to patent or non-patent exclusivities by conducting studies of the therapeutic in children.
Generating Antibiotic Incentives Now (GAIN)
The Generating Antibiotic Incentives Now (GAIN) act of 2012 created five-year extensions of exclusivity for qualified infectious disease products. The exclusivity adds five years of additional exclusivity to approved antibacterial and antifungal drugs to prevent generic entry into the market. For example, 10 years of exclusivity was awarded for ceftolozane/tazobactam, which included five-year new chemical entity (NCE) exclusivity plus five-year qualified infectious disease (QIDP) exclusivity.
Any professional in life sciences or biotech needs to have a comprehensive knowledge of all aspects of therapeutics and modalities. Science for Bankers’ premium content covers each modality in depth, and how each modality or therapeutic is reviewed and approved by the FDA. A tour of Science for Bankers is available here.